Das Medizinprodukte-Durchführungsgesetz (MPDG) ersetzt für Medizinprodukte, die unter die Verordnung (EU) 2017/745 fallen (MDR- Medical Device Regulation) seit 26. Mai 2021 und für Medizinprodukte, die unter die Verordnung (EU) 2017/746 fallen (IVDR- In vitro diagnostic Regulation) seit 26. Mai 2022 das Medizinproduktegesetz (MPG).
Definitionen
„klinische Prüfung“ (Art. 2 Nr. 45 MDR)
bezeichnet eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird
„sonstige klinische Prüfung“ (§3 Abs. 4 MPDG)
eines Produktes ist eine klinische Prüfung, die
- nicht Teil eines systematischen und geplanten Prozesses zur Produktentwicklung oder der Produktbeobachtung eines gegenwärtigen oder künftigen Herstellers ist,
- nicht mit dem Ziel durchgeführt wird, die Konformität eines Produktes mit den Anforderungen der Verordnung (EU) 2017/745 nachzuweisen,
- der Beantwortung wissenschaftlicher oder anderer Fragestellungen dient und
- außerhalb eines klinischen Entwicklungsplans nach Anhang XIV Teil A Ziffer 1 Buchstabe a der Verordnung (EU) 2017/745 erfolgt;
„Leistungsstudie“ (Art. 2, Nr. 42 IVDR)
bezeichnet eine Studie zur Feststellung oder Bestätigung der Analyseleistung oder der klinischen Leistung eines Produkts
„interventionelle klinische Leistungsstudie“ (Art. 2 Nr. 46 IVDR)
bezeichnet eine klinische Leistungsstudie, bei der die Testergebnisse Auswirkungen auf Entscheidungen über das Patientenmanagement haben und/oder zur Orientierung der Behandlung verwendet werden
„Medizinprodukt“ (Art. 2 Nr. 1 MDR)
bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper — auch aus Organ-, Blut- und Gewebespenden — stammenden Proben und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung,
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.
„In-vitro-Diagnostikum“ (Art. 2 Nr. 2 IVDR)
bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern
- über physiologische oder pathologische Prozesse oder Zustände,
- über kongenitale körperliche oder geistige Beeinträchtigungen,
- über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
- zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
- über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
- zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Probenbehältnisse gelten als auch In-vitro-Diagnostika
Voraussetzung ist die Erfüllung der Definition einer klinischen Prüfung nach Art. 2, Nr. 45 MDR, siehe oben
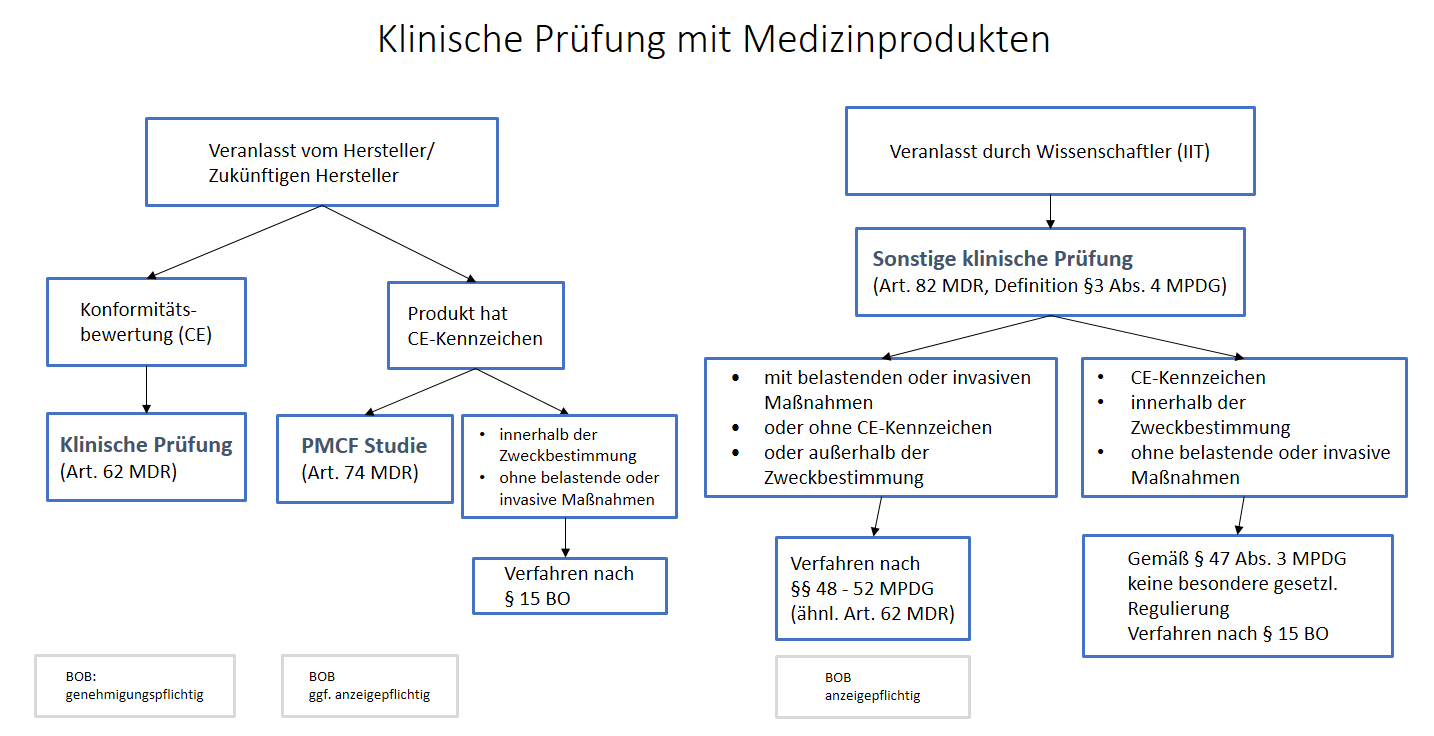
Siehe auch Entscheidungsbaum BfArM und Leitfaden zur regulatorischen Einordnung klinischer Studien mit Medizinprodukten
Siehe Entscheidungsbaum BfArM
Sequenzielles Antragsverfahren
Der Antrag bei der BOB erfolgt erst nach Erhalt der zustimmenden Stellungnahme der Ethik-Kommission.
Antragstellung über das DMIDS
Entsprechend §33 MPDG ist die erforderliche Stellungnahme einer Ethik-Kommission über das Deutsche Medizinprodukteinformations- und Datenbanksystem (DMIDS) nach § 86 zu beantragen https://www.bfarm.de/DE/Medizinprodukte/Portale/DMIDS/_node
bei
- der nach Landesrecht für den Prüfer zuständigen Ethik-Kommission,
- der nach Landesrecht für den Hauptprüfer zuständigen Ethik-Kommission, (wenn ein Hauptprüfer bestimmt ist), oder
- der nach Landesrecht für den Leiter der klinischen Prüfung zuständigen Ethik-Kommission, (wenn ein Leiter der klinischen Prüfung bestimmt ist)
Der Antrag muss enthalten:
- die Angaben und Unterlagen, die in Anhang XV Kapitel II der MDR bzw. Anhang XIV Kapitel I IVDR genannt sind (mit leichten Unterschieden je nach Unterkategorie der klinischen Prüfung, s. jeweilige Artikel der MDR/IVDR)
- den Namen, die Anschrift und die Kontaktdaten des Leiters der klinischen Prüfung, sofern ein Leiter bestimmt ist.
Unterlagen, die für den Prüfungsteilnehmer bestimmt sind, sowie die Zusammenfassung des klinischen Prüfplans sind in deutscher Sprache einzureichen. Die weiteren Angaben und Unterlagen können in deutscher oder englischer Sprache vorgelegt werden.
Fristen zur Stellungnahme der Ethik-Kommission (gemäß §34, §35 MPDG):
- Die Ethik-Kommission bestätigt innerhalb von 10 Tagen den Eingang des ordnungsgemäßen Antrags
- Wurde der Antrag nicht ordnungsgemäß gestellt (formale Prüfung), reicht der Sponsor fehlende Dokumente innerhalb von 10 Tagen nach.
- Die Ethik-Kommission übermittelt dem Sponsor ihre Stellungnahme innerhalb von 30 Tagen nach Eingang des ordnungsgemäßen Antrags.
- Werden zusätzliche Informationen angefordert (inhaltliche Prüfung), ist der Ablauf der Frist für die EK gehemmt. Der Sponsor übermittelt die zusätzlichen Informationen innerhalb einer von der EK bestimmten Frist (max. 45 Tage).
s. Homepage des AKEK
Klinische Prüfungen nach MPDG
Hat ein Sponsor die Absicht, Änderungen an einer klinischen Prüfung vorzunehmen, die wahrscheinlich wesentliche Auswirkungen auf die Sicherheit, die Gesundheit oder die Rechte der Prüfungsteilnehmer oder die Belastbarkeit oder Zuverlässigkeit der im Rahmen der Prüfung gewonnenen klinischen Daten haben, teilt er innerhalb einer Woche der Ethik-Kommission, die Gründe für die Änderungen und deren Art über das internetbasierten Erfassungssystem des DMIDS mit (Art. 75 MDR bzw. Art. 71 IVDR).
Der Sponsor übermittelt eine aktualisierte Fassung der einschlägigen Unterlagen gemäß Anhang XV Kapitel II MDR bzw. Anhang XIV Kapitel II IVDR als Teil der Mitteilung. Änderungen der einschlägigen Unterlagen müssen eindeutig gekennzeichnet sein.
Die Ethik-Kommission nimmt innerhalb von 38 Tagen nach Eingang des Änderungsantrags dazu Stellung.
Anträge ohne Begründung der Änderungen sind formal unvollständig und können nicht in den Geschäftsgang der Ethik-Kommission aufgenommen werden.
Klinische Prüfungen die entsprechend der Übergangsregelung vor dem 26. Mai 2021 (MDR) bzw. 26.Mai 2022 (IVDR) eingeleitet wurden
Beabsichtigt der Sponsor nach Genehmigung der klinischen Prüfung eine wesentliche Änderung, so beantragt er unter Angabe des Inhalts und der Gründe der Änderung (§ 22 c Abs. 2 MPG) bei der zuständigen Bundesoberbehörde eine Genehmigung und bei der zuständigen Ethik-Kommission eine Bewertung der angezeigten Änderungen.
Als wesentlich gelten insbesondere Änderungen, die sich auf die Sicherheit der Probanden auswirken können, die Auslegung der Dokumente beeinflussen, auf die die Durchführung der klinischen Prüfung gestützt wird, oder die anderen von der Ethik-Kommission beurteilten Anforderungen beeinflussen.
Die Ethik-Kommission nimmt innerhalb von 30 Tagen nach Eingang des Änderungsantrags dazu Stellung.
Anträge ohne Begründung der Änderungen sind formal unvollständig und können nicht in den Geschäftsgang der Ethik-Kommission aufgenommen werden.
Für Forschungsvorhaben mit
(1) CE-gekennzeichneten Medizinprodukten, die
(2) im Rahmen der jeweiligen Zweckbestimmung und
(3) ohne zusätzliche belastende oder invasive Maßnahmen durchgeführt werden,
ist weiterhin eine berufsrechtliche Beratung nach § 15 BO möglich (vergleichbar mit dem nicht mehr anwendbaren § 23b MPG).
Für Forschungsvorhaben mit Medizinprodukten ohne CE-Kennzeichnung, ist ebenfalls eine berufsrechtliche Beratung nach § 15 BO möglich, wenn sie nicht mit dem Zweck durchgeführt werden, die Sicherheit oder Leistung des Produkts zu bewerten.
–> Einreichung über ethikPool
| Art der Medizinproduktestudie | Gebühr | |
| Federführend | beteiligt | |
| Erstantrag | ||
| Klinische Prüfung (Art 62 MDR & 70 Abs. 7 ( a, b) MDR / § 31 (1,2) MPDG)
+ jede weitere Prüfstelle |
3500 €*
200 € |
800 €
|
| PMCF (Art 74 MDR)
+ jede weitere Prüfstelle |
3500 €
200 € |
800 €
|
| Sonstige klinische Prüfung (Art. 82 MDR/ § 47 ff MPDG) | 1500 €* | 400 €* |
| Leistungsstudie (Art. 66 Abs. 7 (a,b) IVDR / §31 a Abs. 1,2 MPDG)
+ jede weitere Prüfstelle |
3500 €
200 € |
800 €
|
| PMPF (Art. 70 Abs. 1 MDR)
+ jede weitere Prüfstelle |
3500 €
200 € |
800 €
|
| Leistungsstudie mit CDx an Restproben (§ 31 b MPDG)
+ jede weitere Prüfstelle |
3500 €
200 € |
800 €
|
| Wesentliche Änderung | ||
| Klinische Prüfung | 800 € | 200 € |
| PMCF | 800 € | 200 € |
| Sonstige klinische Prüfung | 600 €* | 150 €* |
| Leistungsstudie | 800 € | 200 € |
| PMPF | 800 € | 200 € |
| Leistungsstudie mit CDx an Restproben | 800 € | 200 € |
| Kenntnisnahme | 100 € | |
| Einbeziehung externer Sachverständiger | Auslagen werden zusätzlich berechnet | |
* Bei Investigator Initiated Trials kann auf Antrag von den üblichen Gebührensätzen abgewichen werden:
